|
|
Self-limiting polymerization of DNA origami subunits with strain accumulation
Jonathan F. Berengut, Chak Kui Wong, Julian C. Berengut, Jonathan P.K. Doye, Thomas E. Ouldridge and Lawrence K. Lee
ACS Nano 14, 17428–17441 (2020)
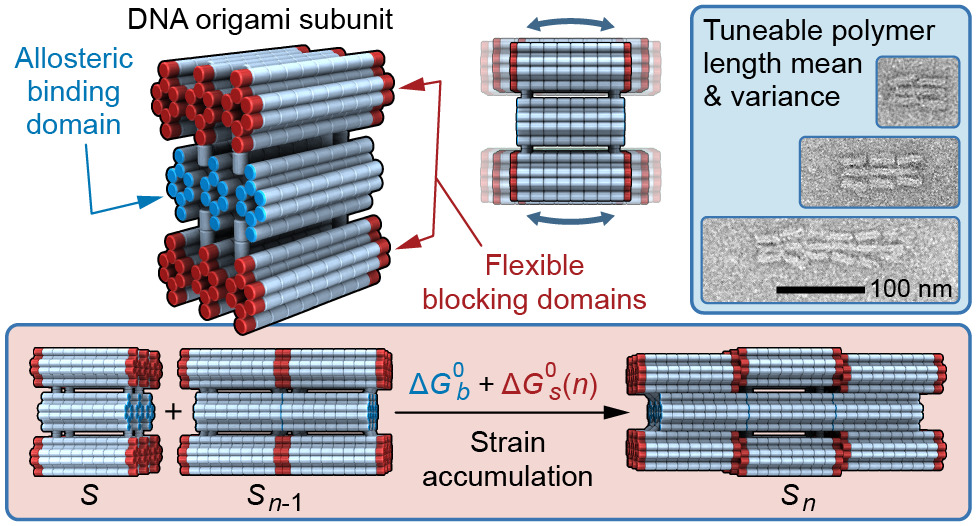
Abstract
Biology demonstrates how a near infinite array of complex systems and structures at many scales can originate from the self-assembly of component parts on the nanoscale. But to fully exploit the benefits of self-assembly for nanotechnology, a crucial challenge remains: How do we rationally encode well-defined global architectures in subunits that are much smaller than their assemblies? Strain accumulation via geometric frustration is one mechanism that has been used to explain the self-assembly of global architectures in diverse and complex systems a posteriori. Here we take the next step and use strain accumulation as a rational design principle to control the length distributions of self-assembling polymers. We use the DNA origami method to design and synthesise a molecular subunit known as the PolyBrick, which perturbs its shape in response to local interactions via flexible allosteric blocking domains. These perturbations accumulate at the ends of polymers during growth, until the deformation becomes incompatible with further extension. We demonstrate that the key thermodynamic factors for controlling length distributions are the intersubunit binding free energy and the fundamental strain free energy, both which can be rationally encoded in a PolyBrick subunit. While passive polymerisation yields geometrical distributions, which have the highest statistical length uncertainty for a given mean, the PolyBrick yields polymers that approach Gaussian length distributions whose variance is entirely determined by the strain free energy. We also show how strain accumulation can in principle yield length distributions that become tighter with increasing subunit affinity and approach distributions with uniform polymer lengths. Finally, coarse-grained molecular dynamics and Monte Carlo simulations delineate and quantify the dominant forces influencing strain accumulation in a molecular system. This study constitutes an important fundamental investigation of the use of strain accumulation as a rational design principle in molecular self-assembly.The full paper is available from ACS Nano